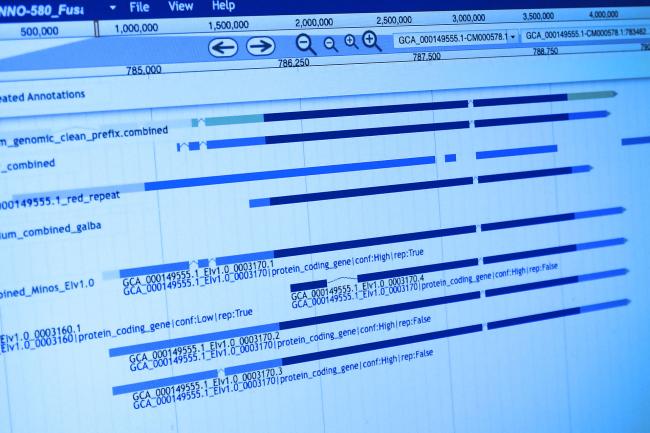
The basis of Ben’s postdoctoral studies, between the Earlham Institute and the UEA, is to build tools and infrastructure in order to study population genomics, so that people can start writing useful population genetics pipelines with Julia.
The most important aspect of this, according to Ben, is design. “It’s not enough to use a ‘fast language’ like C or C++ or Julia, the great thing about julia is that combining ease of comprehension and performance means you can spend more time thinking about the best algorithms to use or implement”.
Ben explains, “Let me give one example where paying attention to data structure and algorithm design pays off: A lot of quickly written (rushed!) scripts just treat DNA as a string of characters, which is not very efficient as one character is represented with one byte. However, you can actually represent every nucleotide with 2 bits [¼ of a byte].
“Imagine A = 00, T = 01, G = 10 and C = 11 (or something like that). So you save space in memory, as 32 nucleotides can be squeezed into a single 64 bit Integer. And then you can do certain operations using bit level operations on that one 64 bit Integer, effectively doing the same operation on 32 nucleotides at the same time in one operation - saving time.
“Compare this with a sloppy implementation, where each nucleotide is stored as one character, than 32 nucleotides requires 32 operations, not 1!”
“It’s surprising sometimes how much you can improve the performance of software and pipelines before you even start to think about multiple cores and parallel programing, although julia makes parallel programming easier too.”
Occasionally, Ben adds, there is a trade-off to make: “Sometimes you have ambiguous characters, such as an N in biological sequences. That means you have a bigger alphabet and so that two bit encoding is not possible.
“However, you can still fit each nucleotide into a “nibble” (4 bits, ½ a byte). Even then, you can fit 16 nucleotides in a single 64 bit Integer and can do operations on 16 nucleotides at once, and this is still a significant speed gain and saving of space.
“Additional speedups can come from the implicit parallelism possible thanks to the Single Instruction Multiple Data (SIMD) capabilities of modern processors.”
With a little julia, and some intelligent thinking about how algorithms are written, the BioJulia team believe it is possible to make bioinformatics software faster, more efficient, and easier to comprehend - giving biologists more time to think about important research questions, rather than writing code.