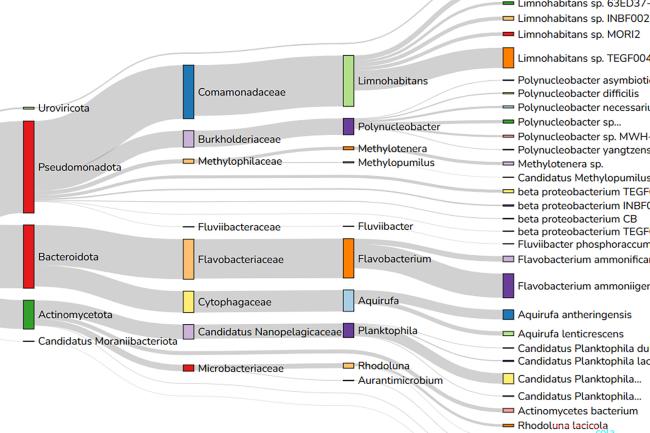
Big data analysis comes with large computing requirements, as well as an ever-increasing range of software and tools. However, not all research groups have equal access to these resources or have the necessary computing specialists to install and run specific programmes and platforms.
The aim of CyVerse is to bring all of this together, allowing researchers to store, access and reuse different datasets, models and analytical tools hosted in an interconnected network of international institutions. CyVerse is a project initiated in the US and developed under a large National Science Foundation (NSF) grant.
The Davey Group at EI are leading the deployment of CyVerse UK - the first CyVerse node outside of the US, which provides free HPC facilities for UK scientists to allow better data analysis and management.
The specific aims of CyVerse are to ensure reproducibility and shareability, with applications running in tandem across various computing environments. Data can be shared at any time either publicly or privately among collaborators, while the proper documentation and code is open source to ensure that others can build on and benefit from CyVerse.
Essentially, if we want to get metaphorical, CyVerse is like an amusement arcade for biologists and bioinformaticians. It brings lots of different tools together into one place, making it easier to analyse data from a variety of sources.
So, rather than having to go to someone’s house to play Street Fighter, then travel to the other side of town to have a go on the Dance Mat - all of the games are readily accessible in one place.
Even better still, the amusement arcade publishes its floor plan, where power sockets are, and lets you share lighting, heating, and who can come in through the front door. So, if someone else has their own machine they want to install and run in the arcade, CyVerse is immediately able to help. This makes CyVerse able to support services all the way from low-level ‘plumbing’ applications, all the way up to user interfaces for data sharing and analysis.
Currently, the CyVerse UK node, developed and maintained by Erik van den Bergh and Alice Minotto, provides access to a number of bioinformatics applications and workflows, including Mikado and Gwasser developed at EI, with several others currently in the pipeline.
“It's great to be developing and supporting an open platform like CyVerse UK, especially at the Earlham Institute where we can contribute many tools and resources to the scientific community. It's a great feeling to be able to help more scientists to use the amount of biological data available for their research,” says Alice.
Eventually, we hope that CyVerse UK will become a National Capability at EI, making it a cornerstone of UK bioscience through providing HPC and storage for collaborative research.